TØ 8 – Evolution og bioinformatik
1 Opgave 1. tRNAs familiealbum
Vi skal nu kigge på den bredere evolution af tRNA ved at finde en alignment af alle kendte tRNA-sekvenser i RNA familie-databasen (Rfam).
1.1 Undersøg tRNA-bevarelse i Rfam
Gå til Rfam-databasen og søg efter “tRNA”. Tryk på “Secondary structure” under tRNA-familien. Klik på “seqcons” (sekvensbevarelse) og bpcons (baseparbevarelse). Hvad er mest bevaret, struktur eller sekvens?
I nedre venstre hjørne ses et farvespektrum fra ikke bevaret (0) til stærkt bevaret (1).
1.2 Analyser Rchie-plot for tRNA
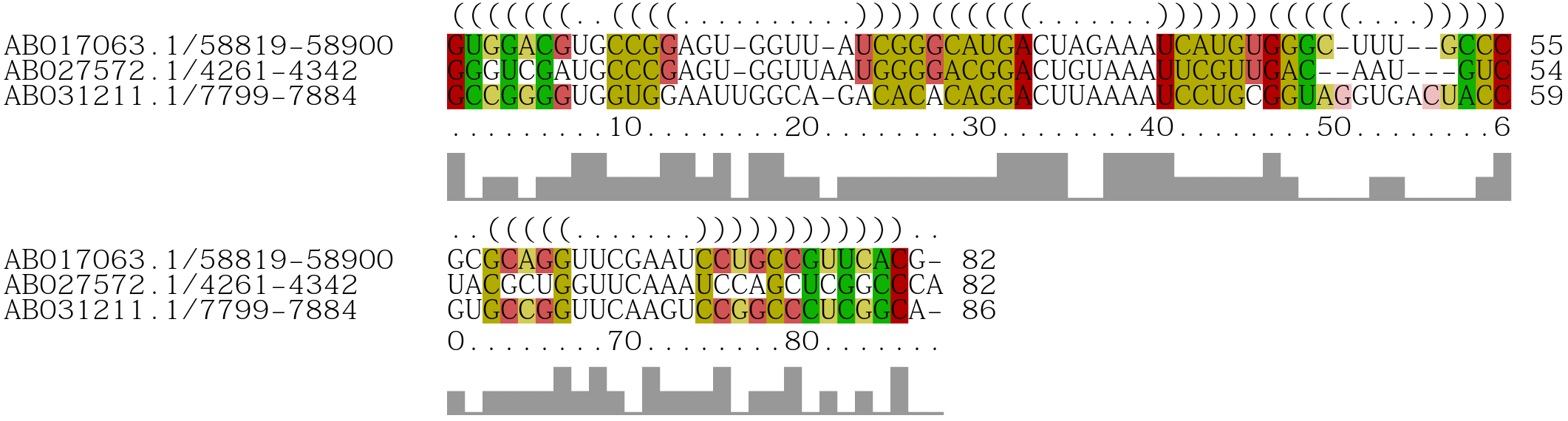
Kig nu på “Rchie” plottet, der vises i boksen herunder. Plottet viser sekvenser (blok med farver, hvor hver vandrette linje repræsenterer en sekvens) og deres sekundær struktur (buer vist på toppen, der forbinder baser der danner basepar). Hvilke stems indeholder mest covariation? Kig ned gennem alignmenten – hvad adskiller nogle tRNA sekvenser fra andre?
Læs om Rchie plots i boksen herunder.
Om de forskellige nukleotidfarver som skrevet i Rchie plots (fra e-RNA.org)
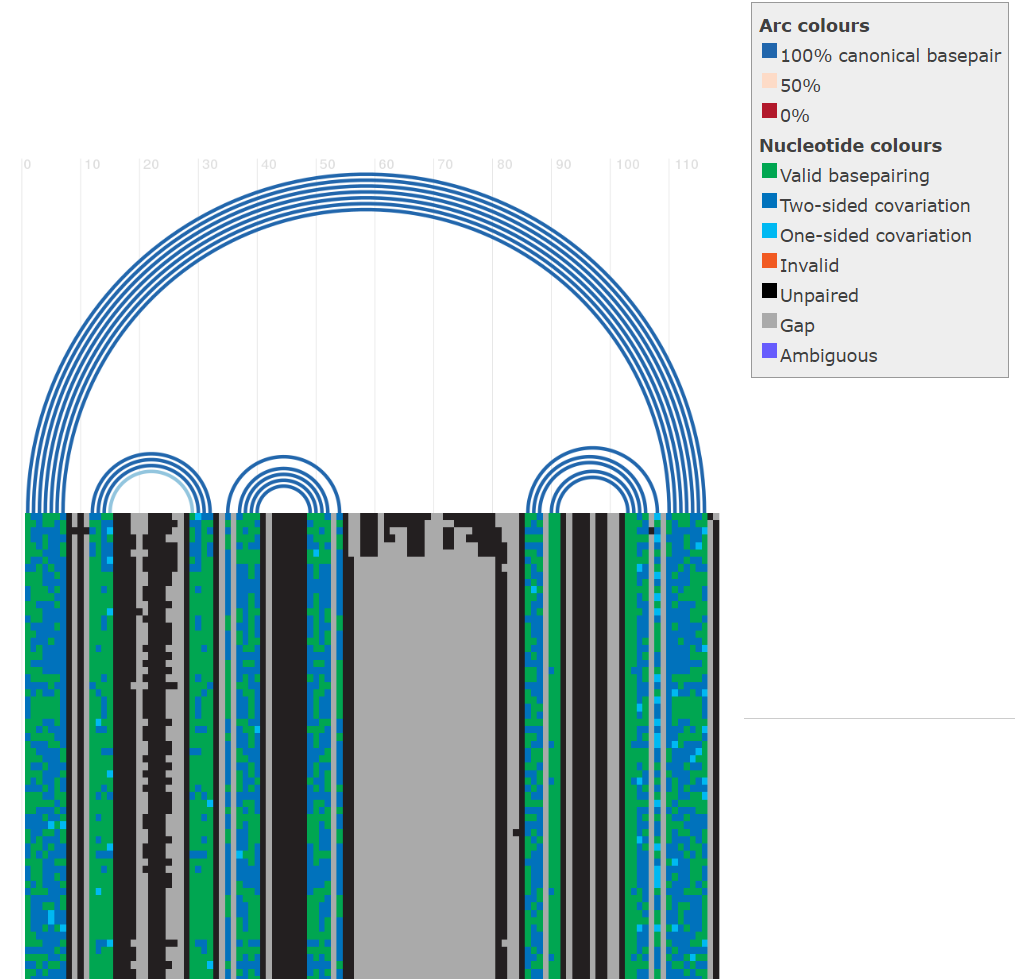
Covariation (correlated variation): refererer til ethvert validt basepar (A:U, G:C, G:U) som adskiller sig fra det mest gængse observerede valide basepar. Det vil sige at der er kompensatoriske baseparændringer (mutationer), som stadig muliggør baseparring samt sekundær struktur.
One-sided covariation: Den ene base i baseparret skifter, men baseparret kan stadig dannes, hvilket er muligt grundet GU wobble. Altså vi har en ændring fra G:C til G:U, fra A:U til G:U eller vice versa.
Two-sided covariation: Begge baser i baseparret adskiller sig fra det mest gængse observerede basepar på denne position. Her er der mange muligheder. Kan både være transitioner og transversioner.
Invalid: alle basepar der ikke er enten A:U, G:C eller G:U
Ambiguous: Enhver base, som ikke er A, C, G, T eller U.
Unpaired: De uparrede. Loops eller bulges.
Vi skal nu undersøge de tRNA sekvenser der adskiller sig. Følgende tRNA-sekvenser er downloadet fra Rfam:
>AB017063.1/58819-58900
GUGGACGUGCCGGAGU-GGUU-AUCGGGCAUGACUAGAAAUCAUGUGGGC-UUU--GCCCG-CGCAGGUUCGAAUCCUGCCGUUCACG
>AB027572.1/4261-4342
GGGUCGAUGCCCGAGU-GGUUAAUGGGGACGGACUGUAAAUUCGUUGAC--AAU---GUCUACGCUGGUUCAAAUCCAGCUCGGCCCA
>AB031211.1/7799-7884
GCCGGGGUGGUGGAAUUGGCA-GACACACAGGACUUAAAAUCCUGCGGUAGGUGACUACCG-UGCCGGUUCAAGUCCGGCCCUCGGCA1.3 Fold atypiske sekvenser med RNAalifold
Prøv at folde dem i RNAalifold server. Hvordan adskiller strukturen sig fra tRNA? Er denne struktur understøttet af co-variationer?
Der ses en masse undermenuer, hvor man kan vælge foldningsalgoritme mm. Disse skal bare ignoreres. De sorte basepar betyder mismatches i alle sekvenser.
Læs om RNAalifold output i boksen herunder.
Én ting der her er værd at bemærke, er at sekvensen, der vises i sekundærstrukturen, er et gennemsnit af de tre alignede, hvor den mest forekomne base typisk er den der vises.
Dette er også grunden til at der ses nogle atypiske basepar (f.eks. C:A, G:G og C:C), men se bort fra det.
>seq1 GUGGACGUGCCGGAGU-GGUU-AUCGGGCAUGACUAGAAAUCAUGUGGGC-UUU--GCCCGCGCAGGUUCGAAUCCUGCCGUUCACG-
>seq2 GGGUCGAUGCCCGAGU-GGUUAAUGGGGACGGACUGUAAAUUCGUUGAC--AAU---GUCUACGCUGGUUCAAAUCCAGCUCGGCCCA
>seq3 GCCGGGGUGGUGGAAUUGGCA-GACACACAGGACUUAAAAUCCUGCGGUAGGUGACUACCGUGCCGGUUCAAGUCCGGCCCUCGGCA-
>output GCGGAGGUGCCGGAGU-GGUU-AUCGGGCAGGACUAAAAAUCCUGUGGC--AUU---ACCGAGCCGGUUCAAAUCCCGCCCUCGACA-
(((((((..((((..........))))((((((.......))))))(((((....)))))..(((((.......))))))))))))..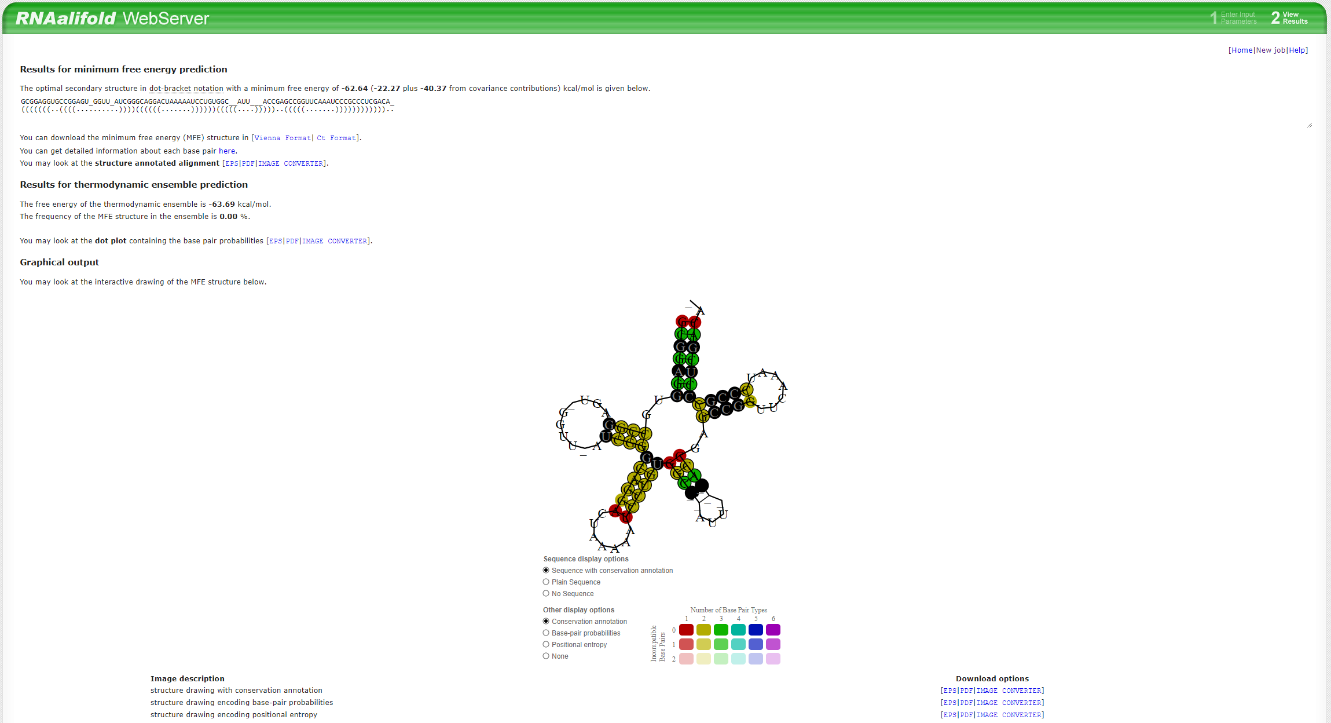
De sorte basepar på strukturen vist under Graphical output, betyder at der kan forekomme inkompatible basepar (mismatches) på denne position i mindst én af sekvenserne og ikke nødvendigvis at der er mismatches i alle sekvenserne.
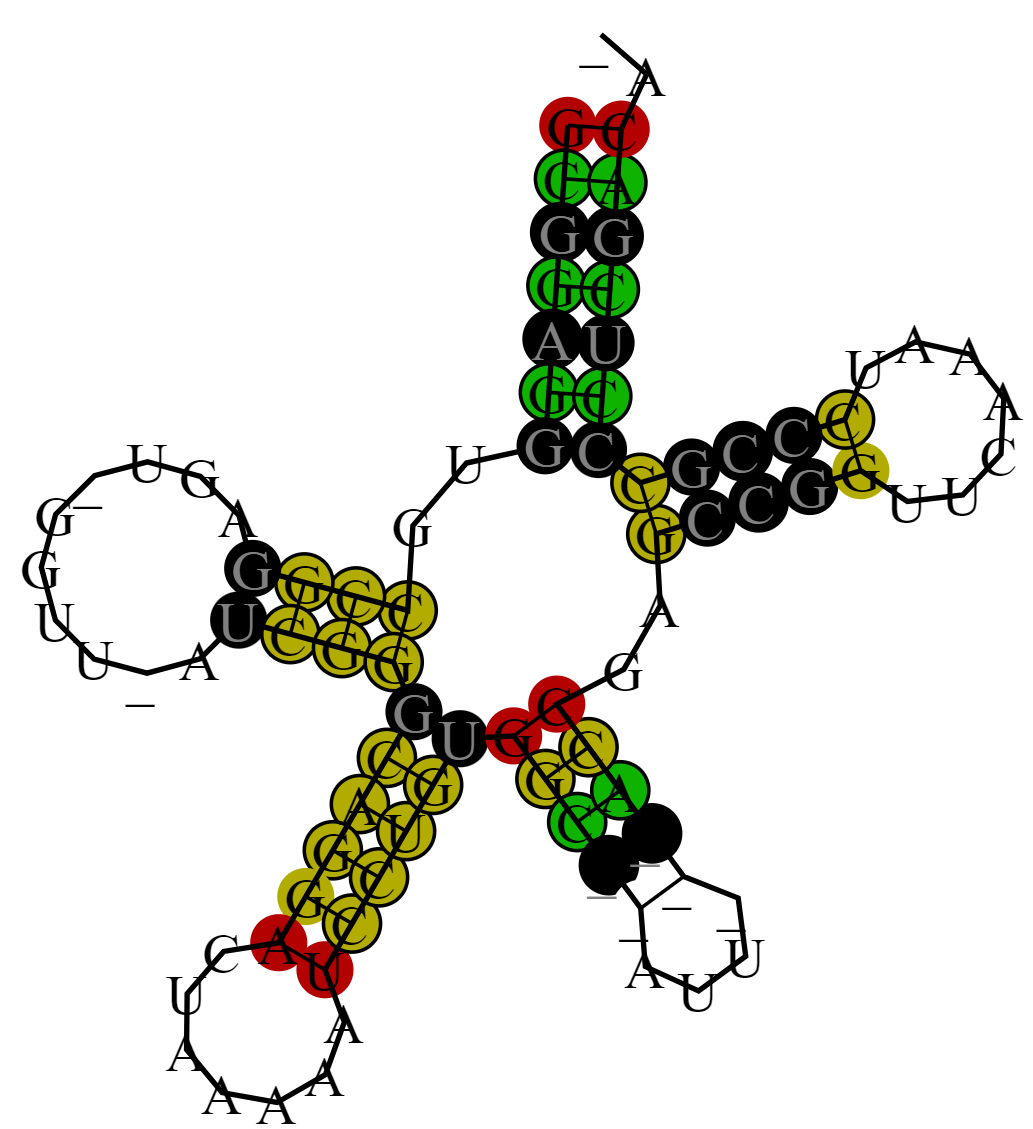
En bedre og mere detaljeret præsentation af tRNA-femkløveren kan ses ved at vælge PDF under Download options ud for ’structure drawing with conservation annotation’ under Image description, som det ses herunder:
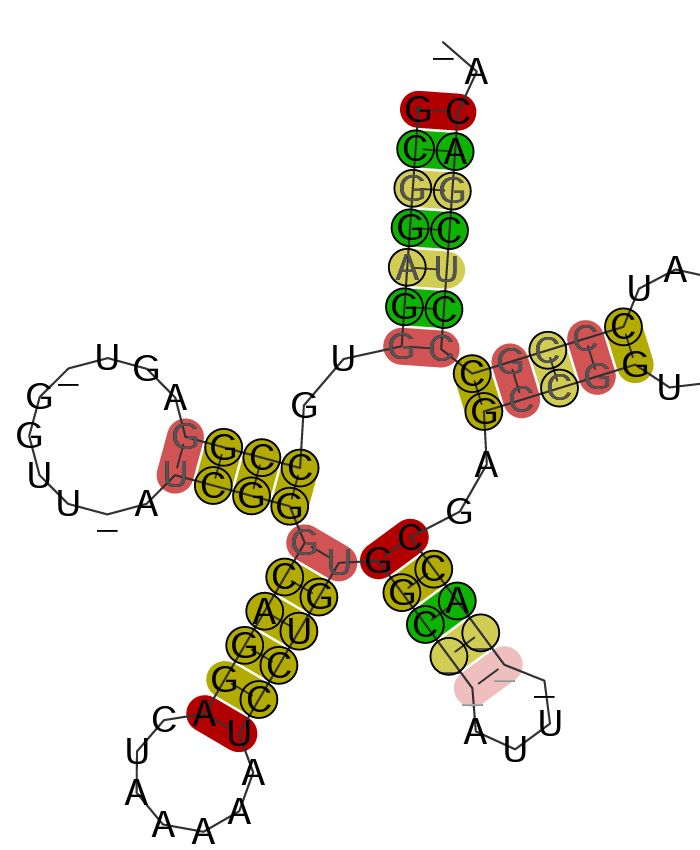
Herved fås en repræsentation med farvning efter antal af forskellige basepar på hver position samt en graduering i gennemsigtighed efter antallet af inkompatible basepar.
Denne kan ydermere akkompagneres med alignment i samme farvekodning og dot-bracket notation ved at vælge PDF ud for structure annotated alignment.
1.4 Sammenlign selenocysteine tRNA-struktur
Selenocysteine tRNA har en lignende struktur. Kig på tRNA-Sec familie på Rfam og sammenlign R-chie plottet. Undersøg nu hvordan den 3-dimensionelle struktur af dette tRNA ser ud. Klik på “Structures” menu-punktet og klik på en af PDB ID’erne (f.eks. 3a3a), der tager dig til PDB. Åben strukturen i PyMOL med fetch 3a3a. Hvor sidder den extra hairpin på den klassiske L-form af tRNA?
2 Opgave 2. Den forsvundne RNA replikase
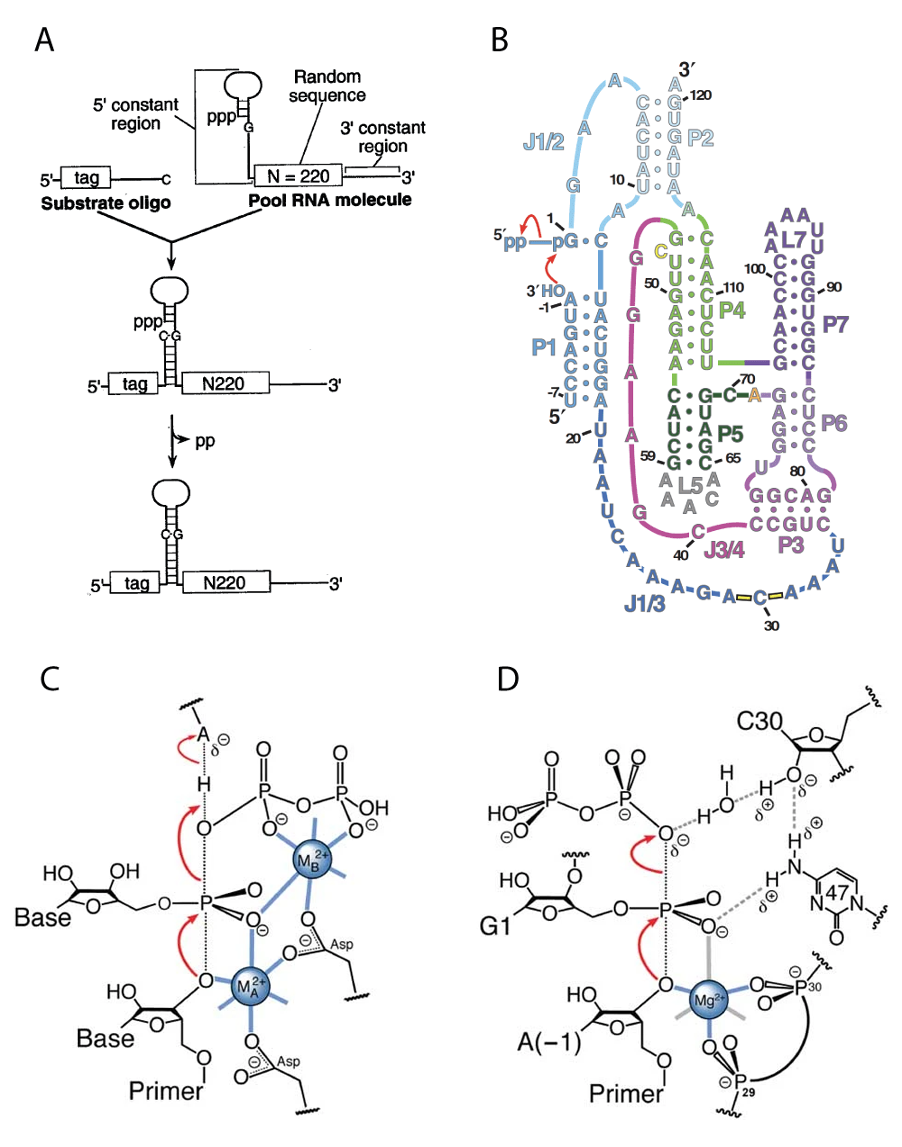
For at sandsynliggøre “RNA world” hypotesen prøvede forskere omkring 1993 at skabe et RNA polymerase ribozym (Bartel & Szostak, 1993) ved at bruge en selektionsmetode, hvor man laver et bibliotek af sekvenser og selekterer for funktion (se Berg, udgave 10, afsnit 10.4). Ved at selektere for ligeringsaktivitet fandt de en RNA ligase, der senere er blevet udviklet til at syntetisere længere RNA-sekvenser (Horning & Joyce, 2016). Til selektionseksperimentet blev der indsat en tilfældig sekvens (et bibliotek) på 220 nukleotider, der syntetiseres kemisk ved tilfældigt at indsætte en af de fire baser på hver af 220 positioner. Figur A viser en oversigt over dette eksperiment, hvor den tilfældige sekvens er annoteret som N220.
2.1 Beregn RNA-biblioteksstørrelse og masse
Hvor meget RNA skal du have i gram hvis biblioteket skal have mindst et molekyle med hver sin sekvens? I det oprindelige eksperiment blev der brugt et bibliotek med 1,6 * 1015 forskellige sekvenser. Hvor mange gram skal bruges til dette bibliotek? Hvad betyder det at de fandt en RNA ligase med dette bibliotek for sandsynligheden for at RNA ligaser kan udvikles?
Den gennemsnitlige molekylevægt for et nukleotid er 330 g/mol og Avogadros tal er 6,0221415 * 1023 mol-1.
2.2 Forudsig konsensus sekundær struktur
Efter ti runder af selektion og amplifikation blev biblioteket sekventeret og der blev fundet flere forskellige familier af sekvenser med ligaseaktivitet. En af familierne (klasse I), der havde god ligeringsaktivitet, er vist herunder som en sekvensaligment.
>b1-10
GGAACACUAUCCGACUGGCACCGUAGAAUACAAAUGUGCCCUCAGAGCUUGGGAAGAUCCUUGCAGGAUCCAGGGGAGGCACCCCCCGGUGGCUUUAACGCCAACGUUCUCAACAAUAGUGGA---
>b1-105
GGAACACUCUACGACUGGAACCGAAAAAUACAAAUGUGCCCUUAGAGCUUGAUAAGAUCCUCGCAGGAUCCAGGGGAGGCACCUCCCGGUGGCUUUAACGCCAACGUUAUCAACAAGAGUGAGAAU
>b1-116
GGAAGACCAUACGACUGGCACCGUACAAUACAAAUGUGCCCUCAGAGCUUGAGAAGAUCCUUGCAGGAUAAAGGGGAGGCACCCCCCGGUAGCUUAAAAGCCAACGUUCUCAACAAUGGUC-ACAABrug RNAalifold webserver til at forudsige konsensus sekundær struktur for RNA ligase klasse I. Sammenlign den forudsagte sekundær struktur med strukturen som forskerne kom frem til (Figur B). Hvilke stem-regioner (P) er forudsagt korrekt? Hvorfor tror du pseudoknuden P3 ikke er forudsagt?
2.3 Identificer kompenserende baseændringer
Hvilke kompenserende baseændringer bliver observeret og hvilke stem-regioner understøtter de? Hvilke stem-regioner modsiges af inkompatible basepar?
2.4 Bestem hvilke stems stacker i PyMOL
I 2009 blev strukturen af RNA ligase klasse I i “post-ligation-state” bestemt med røntgen krystallografi (Shechner et al., 2009) og i 2011 blev den bestemt i “pre-ligation-state” (Shechner & Bartel, 2011). Åben scriptet RNA-ligase.pml i PyMOL. F1 viser RNA strukturen med samme farver som vist i nedenstående figur i panel B - dog er pseudoknuden P3 markeret i rød i PyMOL.
Hvilke stems stacker på hinanden? Hint: To stems kan tilsammen danne en længere helix ved at stacke på hinanden (som det ses f.eks. for P6 og P7 i Figur B).
2.5 Analyser aktive site i pre- og post-ligation
F2 viser active site i “pre-ligation-stateog **F3**post-ligation-state”. Mellem hvilke nukleotider dannes den nye binding? Hvorfor er C47 mon udskiftet med U47 i “pre-ligation-state”? Hvad er C47s rolle i katalysen og hvilken interaktion har C47 med C30?.
Se Figur D
2.6 Sammenlign reaktionsmekanismer RNA og protein
Active site for RNA polymerase proteinet (Figur C) ligner meget active site for RNA ligasen (Figur D). Hvilken reaktionsmekanisme sker når to nukelotider hæftes sammen? Hvilken rolle spiller Mg2+-ion, C30, C47 og vand-molekylet?
3 Opgave 3. Globin sekvensalignment
Hæmoglobin alpha (HGA) og Myoglobin (MYG) fra menneske og Leghæmoglobin-1 (LHG) fra blomsten Lupin er fremhævet som eksempel i Berg Biochemistry, udgave 10, kapitel 10. Følgende sekvenser er hentet fra UniProt i fasta-format.
>HGA
VLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHFDLSHGSAQVKGHGKKVADALTNAV
AHVDDMPNALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTPAVHASLDKFLASVSTVLTSKY
R
>MYG
MGLSDGEWQLVLNVWGKVEADIPGHGQEVLIRLFKGHPETLEKFDKFKHLKSEDEMKASEDLKKHGATVL
TALGGILKKKGHHEAEIKPLAQSHATKHKIPVKYLEFISECIIQVLQSKHPGDFGADAQGAMNKALELFR
KDMASNYKELGFQG
>LHG
MGVLTDVQVALVKSSFEEFNANIPKNTHRFFTLVLEIAPGAKDLFSFLKGSSEVPQNNPDLQAHAGKVFK
LTYEAAIQLQVNGAVASDATLKSLGSVHVSKGVVDAHFPVVKEAILKTIKEVVGDKWSEELNTAWTIAYD
ELAIIIKKEMKDAA3.1 Aligner globiner med ClustalO
Brug ClustalO til at lave en sekvensalignment af Hæmoglobin, Myoglobin og Leghæmoglobin. Hvor mange identiteter er der mellem de tre sekvenser i den forudsagte sekvensalignment?
3.2 Aligner globiner med MUSCLE
Brug MUSCLE til at lave en sekvensalignment af Hæmoglobin, Myoglobin og Leghæmoglobin. Hvor mange identiteter er der mellem de tre sekvenser i den forudsagte sekvensalignment?
3.3 Sammenlign ClustalO og MUSCLE-resultater
Sammenlign ClustalO og MUSCLE. Hvilke forskelle i alignment viser de?
3.4 Find tættest beslægtede globinsekvenser
Klik på “Result Files” og på “Percent Identity Matrix”. Hvilke to af de tre sekvenser er tættest beslægtet og med hvilken identitetsprocent?
3.5 Beregn BLOSUM-62-score for alignment
Protein alignment programmer som MUSCLE bruger Blossum-62 matricen til at score sekvensalignment. Blossum-62 matricen er vist i grafisk repræsentation i Berg Biochemistry, udgave 10, kapitel 10, figur 10.7.
Brug figur 10.7 til at beregne score for de første (fra N-term) af hver slags ændring hhv. “conserved” (markeret med * tegn), “conservative” (markeret med : tegn) og “semi-conservative” (markeret med . tegn) i MUSCLE alignment af Hæmoglobin, Myoglobin og Leghæmoglobin.
For tre sekvenser summeres scores for alle sammenlignings-kombinationer.
3.6 Identificer aminosyreegenskaber i BLOSUM-62
Hvilke aminosyreegenskaber er involveret i Blossum-62 score?
4 Opgave 4. Globin strukturalignment
PyMOL-scripting opgave: I denne opgave skal i lære at bruge kommandoerne align og super.
Åben nu pml filen Globin.pml i PyMOL, hvor F1 viser en strukturel alignment af Hæmoglobin alfa kæde (PDB-ID: 1HBB), Myoglobin (PDB-ID: 1MBD) og Leghæmoglobin (PDB-ID: 1GDJ).
4.1 Beregn RMSD med super-kommando
Forklar hvordan PyMOLs super kommando fungerer og brug funktionen til at udregne root mean square deviation (RMSD) mellem 1HBB, 1MBD og 1GDJ.
4.2 Beregn RMSD med align-kommando
Forklar hvordan PyMOLs align(https://pymolwiki.org/index.php/Align) kommando fungerer og brug funktionen til at udregne root mean square deviation (RMSD) mellem 1HBB,1MBDog1GDJ`.
4.3 Sammenlign super og align-kommandoer
I hvilke tilfælde fungerer super og align bedst? Stemmer RMSD generelt overens med “percent identity” fra forrige opgave?
F2 viser identiteter fra forrige opgave i grøn og hæmgruppe i rød.
4.4 Beskriv placering af bevarede aminosyrer
Beskriv overordnet hvor de bevarede aminosyrer er placeret i globin-strukturen som vist i F2. Hvad er afstanden af bindingen mellem Histidin-87 og jern-atomet? Hvorfor vises denne vigtige binding ikke i PyMOL? Hvordan passer denne længde med en kovalent binding overfor en hydrogen-binding?
5 Opgave 5. Globin strukturbevarelse
📄 Download globin-aln-fasta.txt
PyMOL-scripting opgave: I denne PyMOL opgave skal I lære at plotte sekvensbevarelse på protein struktur vha. et Python script, der hedder colCons.py.
Programmet colCons tager en alignment i fasta-format og en atomar struktur som input, beregner sekvensbevarelse ihht. BLOSSUM-62-matricen, og farvelægger hver aminosyre efter en farveskala.
Download følgende zip-fil der indeholder colCons Python-programmet: colCons.zip. Beskrivelse af hvordan programmet fungerer kan læses i toppen af Python-scriptet. allSorts.inp indeholder PyMOL-kommandoer til at køre programmet på to medfølgende eksempler: EFTU og Sortilin.
I denne opgave bruger vi alignment af Hæmoglobin, Myoglobin og Leghæmoglobin fra opgave 4 som eksempel.
Download
globin-aln-fasta.txtog placér filen i colCons mappen.Åben nu PyMOL, navigér til colCons mappen, sæt PyMOL-parametre, og aktivér colCons.py scriptet:
reinitialize
cd [inset folder path]
bg_color white
set opaque_background, off
run colCons.py cd-kommandoen står for “change directory”. Når man bruger cd-kommandoen skal man være opmærksom på hvor i computeren man er på et givent tidspunkt. Man kan tjekke hvor man er med kommandoen pwd (print working directory), der vil give den nuværende sti til PyMOL sessionen. Brug ls-kommandoen for at opliste indholdet af den nuværende mappe. For at komme ind i den rigtige mappe kan man starte sin folder path med cd /, hvilket fører dig til root directory. Herefter indsættes vejen gennem foldere til filen. Man kan se filens folder path ved at højreklikke på filen og trykke “get infopå mac ellerproperties/egenskaber” på windows. Det kunne f.eks. se sådan ud; cd /: Users/JensJensen/Desktop/BioMolStrFunk/TØ/TØ7.*
- Hent nu Myoglobin og brug
colConstil at farvelægge sekvensbevarelse i “rainbow” farveskala fra blå (sat til 50% sekvensbevarelse) til rød (sat til 100% sekvensbevarelse) ihht. globin-aln.fasta:
fetch 1mbd
color_cons('1mbd','globin-aln-fasta.txt',1,1,0.5,1.0,'rain','yellow','red','BLOSUM62',True)5.1 Identificer bevarede aminosyrer ved hæmgruppen
Vis proteinet med cartoon- og stick-repræsentation og svar på følgende spørgsmål.
Hvilke 100% bevarede aminosyrer binder til hæmgruppen? Hvilke aminosyrer kontakter det centrale Fe atom i hæmgruppen? Hvilken aminosyre er “proximal histidine” og hvilken er “distal histidine” (svar på spørgsmål ud fra information i Berg, udgave 10, afsnit 3.2)?
find finder ikke interaktion med Fe, så brug i stedet distance funktionen.
5.2 Analyser overfladebevarelse i myoglobin
Vis proteinet med sphere-repræsentation og svar på følgende spørgsmål.
Hvordan er aminosyrer på overfladen af myoglobin bevaret? Hvordan ser bevarelsen ud omkring bindingssite for hæmgruppen?
5.3 Sammenlign 3D-placering af bevarede rester
Åben nu alle globinerne og farvelæg dem med colCons:
reinit
fetch 1hbb 1gdj 1mbd, async=0
remove solvent
remove not(chain A)
align 1hbb, 1mbd
align 1gdj, 1mbd
reset
color_cons('1hbb','globin-aln-fasta.txt',3,1,0.5,1.0,'rain','yellow','red','BLOSUM62',True)
color_cons('1mbd','globin-aln-fasta.txt',1,1,0.5,1.0,'rain','yellow','red','BLOSUM62',True)
color_cons('1gdj','globin-aln-fasta.txt',2,1,0.5,1.0,'rain','yellow','red','BLOSUM62',True)Vis proteinerne med cartoon- og stick-repræsentation og svar på følgende spørgsmål.
Er 3D placeringen af de 100% bevarede aminosyrer helt ens imellem de tre globiner?
5.4 Find myoglobins strukturelle insert
Myoglobin har et insert på position 57-61 i forhold til Hæmoglobin. Hvilken sekundær struktur har dette insert og hvilken ligand er bundet til denne struktur?
5.5 Sammenlign myoglobin og hæmoglobin strukturelt
Hvad er den største strukturelle forskel mellem Hæmoglobin (1HHB) og Myoglobin (1MBD)?
5.6 Sammenlign hæmoglobin og leghæmoglobin
Hvad er den største strukturelle forskel mellem Hæmoglobin (1HHB) og Leghæmoglobin (1GDJ)?
6 Opgave 6. Heat shock protein 70 evolution
PyMOL scripting opgave: I denne PyMOL opgave skal I lave en strukturel alignment imellem homologe proteiner og bruge informationer fra Interpro databasen til at analysere evolutionære forskelle.
70 kilodalton heat shock proteiner (Hsp70 eller DnaK) og proteiner med lignende struktur eksisterer i stort set alle levende organismer. Hsp70 er en vigtig del af cellens proteinfoldningsmaskineri og hjælper med at beskytte celler mod stress. Start med at finde Hsp70 familien i InterPro databasen.
6.1 Sammenlign HSP74 og HSP7F i alignment
Kig på alignment (klik på Alignments, vælg derefter seed) og sammenlign HSP74 fra menneske og HSP7F fra gær. Hvad er den største forskel mellem dem?
6.2 Skriv PyMOL-script til strukturalignment
Lav et PyMOL-script, der gør følgende:
Åben HSP7F med PDB-ID: 2QXL. Hint: Sørg for kun at vise én monomer. Gem som F1.
Åben DNAK fra E. coli med PDB-ID: 4B9Q. Hint: Sørg for kun at vise én monomer.
Align HSP7F og DNAK. Gem som F2.
Åben HSP med PDB-ID: 1ATR.
Åben Actin fra E. coli med PDB-ID: 1ATN.
Align HSP og Actin.
Gem som F3.
6.3 Lokalisér insert i HSP7F-struktur
Tryk på F1 og identificer positionen for insertet FKKVTKTVKKDDLTIVAHTFGLDAKKLNE (rest 521-549) i HSP7F fra gær. I hvilken sekundær struktur sidder dette insert? Foreslå evolutionær mekanisme hvormed dette insert er blevet dannet.
6.4 Beskriv strukturforskelle i HSP7F og DnaK
Tryk på F2 for at se strukturel alignment af HSP7F og DNAK. Hvilke strukturelle forskelle er der på disse strukturer?
6.5 Beskriv domænestruktur i HSP og actin
Tryk på F3 for at se strukturel alignment af HSP og Actin og beskriv den overordnede struktur af domæner med beta sheets og alfa helices. Er actin og Hsp70 homologe proteiner når de har en forskellig funktion?