TØ 14 – Strukturmetoder
1 Opgave 1. Test dig selv i PyMOL-scripting
Åbn først den vedhæftede fil pymol_template.pml, som kan downloades med knappen ovenfor, i Visual Studio Code. Denne indeholder et template-script, der henter to strukturer af den bakterielle leucine transporter, LeuT til objekterne LeuT1 og LeuT2. Brug scriptet til at besvare opgaven ved at fylde jeres egne kommandoer ind under hvert spørgsmål som angivet i scriptet. Scriptet opretter i alt 8 scener lagret i F1…F8 svarende til hvert spørgsmål. I hvert spørgsmål bygges der videre på scriptet ovenfor, så I skal ikke reinitialize på noget tidspunkt. Når I afleverer scriptet skal hver F-tast vise svaret på det pågældende spørgsmål.
1.1 Vis LeuT1 som cartoon
Vis objektet LeuT1 som “cartoon” med farven lime og sæt en orientering, der viser hele strukturen.
1.2 Selektér Na⁺-ioner som spheres
Opret en selektion med de to Na+-ioner med residue-numre 751 og 752 og vis de to atomer som sphere med farven violetpurple.
1.3 Fremhæv rester 112 og 179
Fremhæv resterne 112 og 179 med sticks og sæt farven sådan at kun carbon er lime mens andre atomer har deres respektive atomfarver.
1.4 Mål afstand som measure-objekt
Angiv den korteste afstand mellem de to rester som et measure-objekt.
1.5 Vis atomer indenfor 6 Å
Opret en anden selektion med alle atomer indenfor 6 Å af de to Na+-ioner. Vis disse som sticks hvor alle atomer ud over carbon farves efter deres respektive atomfarver. Sørg for at intet er selekteret efter scriptet er kørt.
1.6 Align LeuT2 til LeuT1
Vis også strukturen af LeuT2 som “cartoon” og foretag et strukturelt alignment af denne over på LeuT1.
1.7 Farv LeuT2 med elektrostatik
Vis LeuT2 med en overflade farvet efter “vacuum electrostatics”.
2 Opgave 2. Bragg’s lov
2.1 Forklar konstruktiv interferens
Forklar med dine egne ord hvorfor kun bestemte vinkler giver konstruktiv interferens. Brug Braggs lov
\[ 2d \cdot \sin\theta = \lambda \]
til at understøtte din forklaring.
2.2 Sammenlign to proteinkrystaller
Hvis du har to proteinkrystaller:
- Krystal A med større enhedscelle (d = 100 Å mellem proteinlag)
- Krystal B med mindre enhedscelle (d = 50 Å mellem proteinlag)
Hvilken krystal vil give diffraktionspletter længere fra centrum på detektoren? Forklar hvorfor.
2.3 Beregn mindste d-værdi
Ved en bølgelængde af røntgenstrålen \(\lambda = 1.5 \ \mathrm{Å}\).
Hvad er den mindste d-værdi (Bragg opløsning) du kan observere hvis din detektor maksimalt kan måle ved \(\theta = 60^{\circ}\)?
2.4 Forklar krav til høj opløsning
Forklar hvorfor højere opløsning kræver:
- Enten kortere bølgelængde (\(\lambda\))
- Eller evnen til at måle ved større vinkler (\(\theta\))
3 Opgave 3. Røntgenkrystallografi vs. Cryo-EM – Vælg den rette metode
For hvert scenarie nedenfor skal du:
Vælge den mest velegnede metode (Røntgenkrystallografi eller Cryo-EM).
Begrunde dit valg ved at diskutere fordele og ulemper ved begge metoder i den specifikke kontekst.
3.1 Scenarie 1: Lægemiddeludvikling mod en lille, stabil protease
Et medicinalfirma vil udvikle en ny hæmmer mod en viral protease. Proteinet er relativt lille (35 kDa), meget stabilt og kan produceres i store mængder. Målet er at opnå en struktur med den højest mulige opløsning for at kunne se præcis, hvordan lægemiddelkandidaten binder i det aktive site, inklusiv placeringen af enkelte vandmolekyler, der medierer interaktionen.
Hvilken metode vil du anbefale og hvorfor?
3.2 Scenarie 2: Undersøgelse af et stort, dynamisk maskineri
En forskergruppe vil bestemme strukturen af det humane spliceosom (en kæmpe samling af proteiner og RNA på over 1.5 MDa), mens det aktivt splejser et stykke pre-mRNA. Komplekset er kendt for at være meget fleksibelt og eksistere i adskillige forskellige konformationelle tilstande under processen. Det er ekstremt svært at rense komplekset i en helt homogen tilstand.
Hvilken metode vil du anbefale og hvorfor?
3.3 Scenarie 3: Struktur af et membranprotein i sin native membran**
Du vil forstå, hvordan en ionkanal fungerer direkte i cellemembranen, omgivet af lipider og andre membranproteiner. Du er interesseret i at se, hvordan proteinets struktur ser ud in situ (på sin naturlige plads i cellen), ikke som et isoleret, oprenset protein.
Hvilken metode (eller variation af en metode) vil du anbefale og hvorfor?
4 Opgave 4. Modelbygning
📥 Download elektrondensitet.pse
I denne opgave skal vi prøve kræfter med fortolkning af et elektrontæthedskort og modelbygning. Diffraktionsdata for et lille protein på 96 aminosyrerester er blevet indsamlet til en maksimal opløsning på 1.9 Å, og man har beregnet et elektrontæthedskort.
Vi skal kigge på et lille udsnit af elektrontætheden, som kan ses ved at åbne elektrondensitet.pse som kan downloades ovenfor.
Modelbygningen er påbegyndt ved at indsætte en såkaldt poly-Ala-model i tætheden, dvs. en præliminær struktur kun bestående af den mest simple aminosyrerest med en sidekæde, alanin. Modellen kan ses som en gul stick-struktur inde i elektrontæthedskortet, der er vist kontureret med et gitter i blåt.
4.1 Find N- og C-terminal i tætheden
Analysér tæthedskortet og poly-Ala modellen og find peptidkædens N- og C-terminaler. Hvilke to sekundære strukturelementer er dette lille peptidmotiv opbygget af?
4.2 Identificér sidekæder i kortet
Kig nærmere på kortet og se om du kan identificere sidekæder, carbonylgrupper og amidnitrogener i peptidet.
Hint: Dette er lettest at se ved de store sidekæder.
Fra det forudgående molekylærbiologiske kloningsarbejde vides det at peptidsekvensen for fragmentet er følgende:
MNYKELEKMLDVIFENSEIKEIDLFFDFørste trin i modelbygningen er at identificere de store, hydrofobe aminosyrer i sekvensen, da disse ofte er mest ordnede og derfor lettest at finde i tætheden.
4.3 Find hydrofobe aminosyrer i sekvens
Hvilke store, hydrofobe aminosyrer findes i sekvensen og hvor er de placeret? Er der et motiv blandt disse, der kunne tænkes at være let at identificere i kortet?
4.4 Placér motivet i elektrontæthedskort
Find motivet i elektrontæthedskortet baseret på din viden om peptidkædens retning. Passer nogle af de andre aminosyresidekæder?
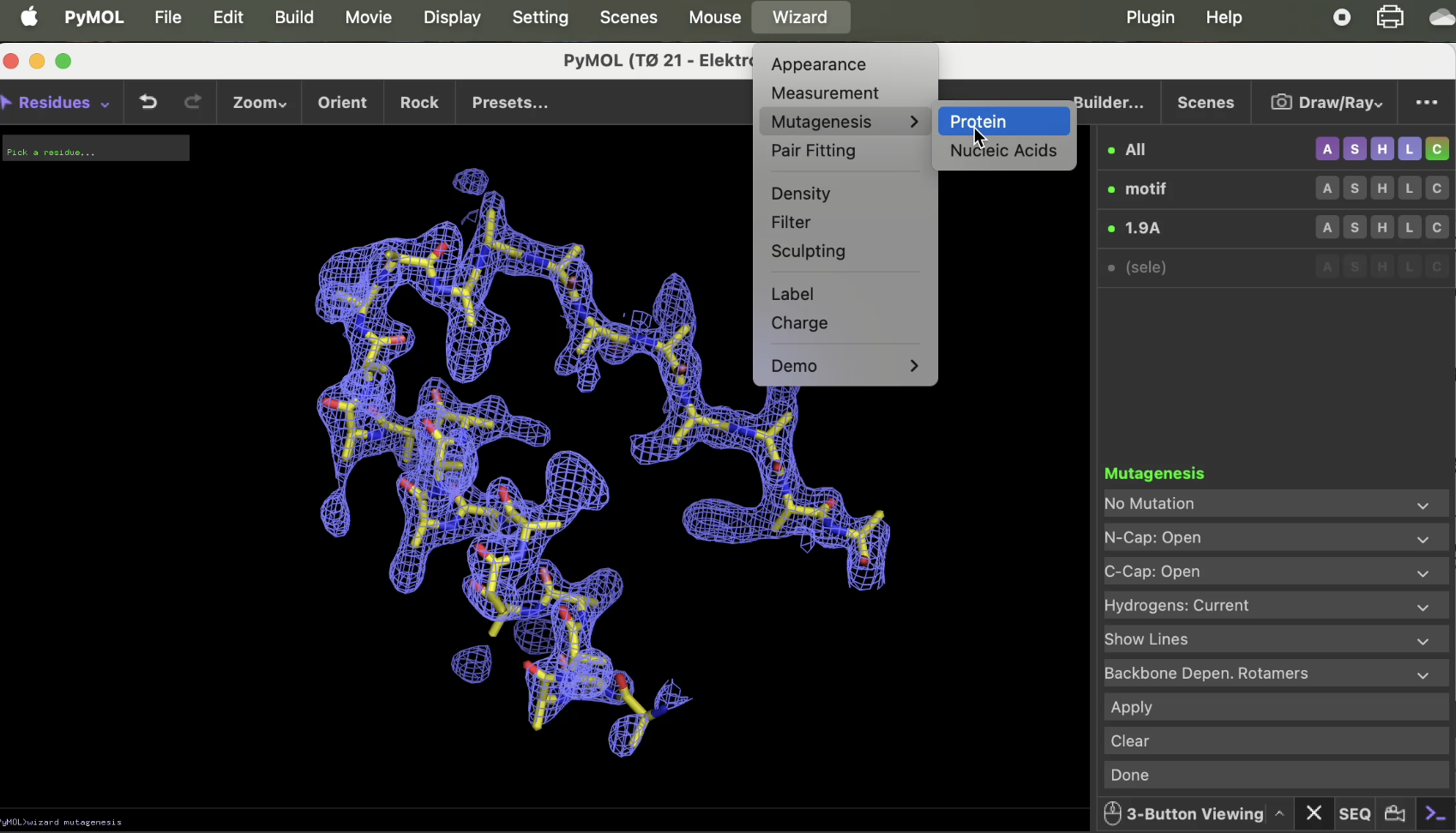
Man kan bruge wizard mutagensis for at redigere på aminosyrer i en peptidkæde. Det kan bruges f.eks. når man skal vurdere effekten af en mutation. Her er det selvfølgelig vigtigt at notere til at proteinet ikke altid vil folde sig præcis på samme måde med en mutation som i vildtypen. Det kan altså kun bruge kvalitativt.
Man trykker på protein under Wizard > mutagenesis, så ses på figuren forneden til venstre, hvorefter menuen på figuren forneden til højre vises. Man selekterer så en enkelt aminosyre, trykker No mutation og trykker så på den ønskede aminosyre. For hver aminosyre bruger man undermenuen rotamer > backbone dependant rotamer. Man trykker så på pilene og går igennem de forskellige konformationer. Når man har fundet en der passer ift. elektrondensitetet. En rød skive indikerer sterisk clash og en grøn skive indikerer stabiliserende interaktioner. Når du har fundet den rigtige, trykker du apply. Et godt tip for at spare tid er at indsætte én aminosyretype ad gangen og at starte med større hydrophobe aminosyrer og derefter større hydrofile aminosyrer. Tryk done når du har ændret alle aminosyrer.
4.5 Vurdér alle aminosyreresternes fit
Kig alle aminosyreresterne igennem og find nogle, der ikke passer så godt i tætheden. Er der en fejl i sekvensen eller hvad kan dette mon skyldes?
Tænk over hvilken type af aminosyrerester, disse tilhører og hvor de normalt findes i et protein. Evt. brug en log-file til at gemme dit arbejde.