TØ 3 - PyMOL-scripting
1 Opgave 1. Selektioner og objekter i PyMOL
Formålet med denne opgave er at forstå forskellen på selektioner og objekter i PyMOL, hvilket er meget vigtigt for at kunne bruge programmet effektivt.
Basalt set er,
objekt: En struktur eller gruppe af atomer inkl. deres position, atomtype, okkupans og temperaturfaktor. Du kan tænke på et objekt som selve strukturen eller PDB-filen. Et objekt dannes vha. kommandoen
create.selektion: Et udvalg af atomer eller kæder i et objekt. Det er vigtigt at forstå at en selektion ikke selv indeholder strukturen, men kun er et udvalg af atomer i et andet objekt. En selektion dannes vha. kommandoen
select
Et objekt kan altså godt eksistere uden en selektion, men en selektion kan ikke findes uden et objekt (i hvert fald ikke med indhold).
1.1 Hent K+-kanalstrukturen 1BL8
Hent strukturen af K+-kanalen fra Streptomyces fra forrige opgave med kommandoen fetch 1BL8. Kommandoer kan enten tastes øverst eller nederst i vinduet hvor der står “PyMOL>”. I denne og følgende opgaver på kurset vil følgende angive en kommando, der skal skrives i PyMOL (eller stå i et script):
fetch 1BL81.2 Dan en selektion af kæde A
Dan en ny selection bestående af kæde A i strukturen:
select select_A, chain AHer er select_A navnet på den nye selektion mens “chain A” fortæller PyMOL at selektionen skal bestå af kæde A. Bemærk at der kommer en ny selektion i listen til højre og at navnet står i parentes, hvilket angiver en selektion. PyMOL viser også selektionen med “selection handles” for hvert atom (små firkanter på strukturen). En selektion fungerer lidt som i andre programmer, du kender, f.eks. forsvinder “selection handles”, hvis man klikker udenfor selektionen. De kommer tilbage ved at klikke på selektionens navn (select_A). Man kan altså hhv. vælge og ikke vælge atomerne ved at klikke på navnet.
1.3 Test selektion- og objektsynlighed
Prøv at vælge selektionen så de små selection handles vises. Sluk nu for hovedobjektet 1BL8 og bemærk at hele strukturen forsvinder, mens selektionen stadig vises. Der er ingen atomer i selektionen, så hvis man slukker for hovedobjektet, ses strukturen ikke.
1.4 Dan et nyt objekt af kæde A
Dan nu et nyt objekt bestående af kæde A i strukturen:
create object_A, chain AHer er objekt_A navnet på det nye objekt mens “chain A” fortæller PyMOL at objektet skal bestå af kæde A. Bemærk at der kommer et nyt objekt i listen til højre og at navnet står uden parentes (og har en 1/1 state), hvilket angiver et objekt. PyMOL centrerer også synsfeltet på det nye objekt.
1.5 Sammenlign objekt og selektion
Ved at oprette et nyt objekt har vi i praksis lavet en kopi af en del af strukturen (atompositioner etc.), så vi nu har to kæde A. Prøv at skjule det oprindelige objekt 1BL8 (klik på objektnavnet) og se at vi stadig kan se den ene kæde i chain_A. Vi har nu to strukturer af kæde A.
Forskellen på objekter og selektioner kan virke pedantisk og unødvendig, men det er vigtigt at du forstår ovenstående og er helt klar på forskellen mellem de to typer i PyMOL. Her er nogle eksempler på hvad de kan bruges til:
selektioner - bruges ofte til at vælge en del af strukturen med henblik på at vise den i en anden repræsentation eller farve.
objekter - bruges til at arbejde uafhængigt med en del af stukturen samt i visse tilfælde hvor man vil vise en struktur i flere repræsentationer samtidig.
1.6 Farv selektion og objekt forskelligt
Prøv at farve selektionen select_A rød og objektet object_A blåt. Tænd og sluk nu for 1BL8 og object_A for at se hvad der er sket. Vi har nu de to kopier af kæde A i to forskellige farver. De er naturligvis perfekt overlejrede, så afhængig af hvilken der sidst har været vist, ses kæden enten som rød eller blå. Vi kan arbejde med de to kopier uafhængigt af hinanden, f.eks. overlejre object_A på en anden struktur, uden at det påvirker 1BL8. Overlejrer vi derimod selektionen select_A vil hele strukturen 1BL8 blive flyttet. Mere om det senere.
Læs mere om selektioner i PyMOL her.
2 Opgave 2. Kæder, residues og atomer i PyMOL
Nu hvor du forstår forskellen på selektioner og objekter, skal vi tale om PyMOL’s generelle selektion kaldet sele. Denne opstår når man klikker med musen på en struktur eller vælger en del af sekvensen ud.
2.1 Hent 1BL8 og ryd PyMOL
Hent strukturen af Streptomyces K+-kanalen som i de forrige opgaver (fetch 1BL8). Har man allerede startet PyMOL og vil slette alt, kan man skrive kommandoen reinit (reinitialize), eller vælge File->Reinitialize->Everything fra menuen.
2.2 Vis 1BL8 som sticks med atomfarver
Vis 1BL8 som sticks og zoom lidt ind med musen (huske at skjule cartoon-repræsentationen først). Farv den med atomfarver med carbon i gult.
2.3 Identificer Phe103 ved museklik
Midt i kanalen findes en stor hydrofob aminosyre, Phe 103, nær en kavitet. Phenylringen er rimelig tydelig. Klik med musen for at identificere aminosyren og vælge den i sele. Hvis du rammer forkert, klik da udenfor strukturen for at slette selektionen igen. Når man klikker på et atom, skriver PyMOL:
You clicked /1BL8/A/A/PHE`103/CG
Selector: selectionseledefined with 11 atoms.
Informationen fortæller os at i objektet 1BL8, segment A, kæde A klikkede vi på residue Phe103 og atomet CG (Cγ). I praksis er segment ID altid lig med kæde ID, så derfor står der A to gange. PDB-formatet understøtter ikke græske tegn, så derfor er atomnavnene oversat til latinske bogstaver:
| Atomnavn | PDB-navn |
|---|---|
| α | A |
| β | B |
| γ | G |
| δ | D |
| ε | E |
| ζ | Z |
| η | H |
2.4 Forstå PDB-atomnomenklatur
Cα hedder altså CA, Oγ hedder OG og så fremdeles. Nogle gange (f.eks. i Asp) kan der være to ens atomer på samme position, hvorved de navngives 1 og 2, f.eks. hedder de to carboxylatatomer i Asp OD1 og OD2.
2.6 Dan selektioner med PyMOL-nomenklatur
Vi kan også bruge nomenklaturen ovenfor til at danne en selektion med CG-atomet fra Phe 103:
select 103CG, /1BL8/A/A/PHE\`103/CGDet lille backquote (`) kan godt drille, men det behøves heldigvis heller ikke:
select 103CG, /1BL8/A/A/103/CGMan behøver heller ikke give alle felterne, hvis de ikke er nødvendige, f.eks. i dette tilfælde er der kun én struktur, så vi kan nøjes med
select 103CG, A/103/CGNår du laver selektioner, så husk at tjekke at det forventede antal atomer vælges ved at se hvad PyMOL skriver:
Selector: selection
103CGdefined with 1 atoms.
2.7 Anvend delvise selektionsstier
Hvis man kun benytter en del af felterne er reglen at hvis udsagnet starter med / så skal man have alt med fra venstre men må godt stoppe halvvejs. Starter udsagnet ikke med / fortolkes det som at starte fra højre. Eksempler:
Vælg hele kæde A (fra venstre):
select chain_A, /1BL8/A/AVælg hele residue 103 i kæde A (fra venstre):
select A103, /1BL8/A/A/103Vælg kun CG i residue 103 (fra højre):
select 103CG, 103/CGHvor mange atomer vælges i dette tilfælde? Hvorfor?
2.8 Vælg residues med udeladt information
En anden nyttig regel er at man kan udelade noget af informationen for at vælge alt, f.eks. vil
select res103, /1BL8///103vælge alle residues i strukturen 1BL8 med residuenummer 103. Hvor mange atomer/residues er det?
Da der kun er ét objekt, kunne vi endda bare sige,
select res103, ////1032.9 Vælg residues i et interval
Man kan også angive en range af residues vha. en bindestreg, f.eks. vil
select A103-105, /1BL8//A/103-105vælge residues 103-105 i kæde A.
2.10 Vælg residues fra proteinsekvensen
Endelig kan man vælge residues i proteinsekvensen. Vælg Display->Sequence fra menuen og træk fra residue Arg27-Leu36 i sekvensen for at vælge det. Find selv ud af hvordan man vælger i de andre kæder (Hint: Sæt først kæderne til forskellige farver, da de vises i sekvensen).
2.12 Visualiser strukturen med selektioner
Brug dine selektioner til at vise hele strukturen i cartoon med en lys grøn farve, farv kæde D mørkegrøn, farv alle Phe-residues i strukturen røde, vis Trp87 i kæde D med sticks og atomet Nε1 med en blå sphere. Vis alle Cα-atomer i kæde A med røde spheres. Farv området 110-120 i kæde B gult.
2.13 Diskuter farveændring ved Cα-farve
Hvad skete der med cartoon-farven i kæde A, da du ændrede farven på Cα-atomerne? Diskutér.
Læs mere om selektioner i PyMOL her.
3 Opgave 3. Avancerede selektioner og boolsk algebra i PyMOL
Nomenklaturen for atom-selektion, som vi arbejdede med i forrige opgave, er nyttig men bliver hurtigt kompleks og svær at læse. Derfor kan man opnå det samme i PyMOL med mere letlæselige kommandoer, hvoraf de vigtigste er,
chain - vælger en kæde
resi - vælger residues efter deres nummer
resn - vælger residues efter deres navn (aminosyre trebogstavkode)
name - vælger atom efter navn
Disse kan kombineres med de boolske operatorer OR, AND og NOT, hvor OR giver foreningsmængden af to selektioner og AND fællesmængden.
3.1 Hent 1BL8 og lav kædeselektioner
Start PyMOL og hent strukturen af Streptomyces K+-kanalen i 1BL8. Lav en selektion af kæde A, som vi gjorde i opgave 1,
select chainA, chain ALav en anden selection med Trp87 i kæde D vha. and-operatoren:
select D87, chain D and resi 87Bemærk at “chain D” vælger alle atomer i kæde D mens resi 87 vælger alle atomer med residuenummer 87. AND giver fællesmængden, hvilket er alle atomer, der både er i kæde D og i residue 87.
Husk som altid at tjekke at du får det forventede antal atomer. PyMOL skriver,
Selector: selection `D87` defined with 14 atoms.14 atomer er som forventet antallet i én Trp. Fint.
3.2 Sammenlign AND og OR operatorer
Mange begyndere bliver ofte usikre på om de skal bruge AND eller OR. Hvad ville resultatet være ved at skrive,
select D87, chain D or resi 87Prøv det og forklar resultatet.
3.3 Vælg atomer med name og resn
På samme måde kan man vælge atomer efter navn (se forrige opgave) med “name” og residues efter navn (f.eks. Trp) vha. “resn”. Skriv nu kommandoer til at lave selektionerne fra før og giv dem fornuftige navne,
- Residues 110-120 i kæde B.
- Alle residues i kæde D.
- Alle Cα-atomer i kæde A.
- Residue Trp87 i kæde D.
- Atomet Nε1 i Trp87 i kæde D.
- Alle Phe-residues i strukturen.
3.4 Kombiner selektioner med OR-operatoren
Foreningsmængde-operatoren OR benyttes når man skal vælge mere end én ting, f.eks. vælges både kæde A og B således,
select chainsAB, chain A or chain BSkal man kombinere AND or OR bliver det vigtigt at benytte parenteser, f.eks. vil udsagnet
select my_res, (chain A and resi 87) or (chain B and resi 110-120)vælge både residue 87 i kæde A og residues 110-120 i kæde B i én selektion.
3.5 Generer specifikke selektioner
Brug nu ovenstående viden til at generere følgende selektioner,
- Residues 10-30 i kæde A og kæde B.
- Atomet Cα i residue 87 i kæde A samt samme atom i residue 86 i kæde B.
- Alle Cys-residues i kæde A og alle Met-residues in kæde B.
3.6 Identificer og farv den lange helix
Vis strukturen som ribbon for at lette identifikationen af Cα-atomer. Identificér start og slut residues for den lange, yderste helix i strukturen, der er én i hver kæde, så det er lige meget hvilken du bruger. Lav en selektion, der indeholder hele den lange helix i alle kæder og farv dem røde.
3.7 Farv strukturen undtagen N-terminalen
Hvis man vil vælge det hele, men undlade en lille del, kan negationsoperatoren NOT være nyttig. F.eks. vil,
select noNterm, 1BL8 and not resi 1-30vælge hele strukturen pånær de N-terminale residues 1-30. Prøv at farve alt undtagen N-terminalen blå.
3.8 Vælg kæde A uden Cys-residues
Lav en selection, der vælger alle residues i kæde A, undtagen Cys-residues.
Læs mere om selektioner i PyMOL her.
4 Opgave 4. Scripting i PyMOL
Scripting dækker over en form for programmering, hvor de enkelte kommandoer fortolkes løbende uden at kildekoden først skal oversættes til en binær fil af en compiler. Det kan også kaldes macro-programmering, da vi skaber et lille program ved at sammensætte en række kommandoer, der udføres én ad gangen.
PyMOL-scripts kan indeholde PyMOL-kommandoer som dem vi har set indtil nu (select, create, show, hide) eller Python-instruktioner, der giver mulighed for meget avancerede scripts med loops, if-then-else statements osv. Python-programmering i PyMOL dækker vi senere på dette kursus, hvor vi inddrager det, I lærer om Python på Bioinformatik-kurset direkte i scripts i PyMOL.
Et PyMOL-script består af en tekstfil med enten .pml (Python macro language) eller .py (python) efternavn. Det anbefales, at du opretter en folder til dine scripts, så du senere kan finde dem frem igen. Scripts kan kaldes direkte inde fra PyMOL vha @-funktionen, men her vil vi starte med at kopiere scriptet og indsætte det på PyMOL’s kommandolinje.
Til redigering af scripts skal du bruge en plain text-editor, som VS Code.
PyMOL-scripts er typisk opbygget på følgende måde:
- Først initialiseres og generelle indstillinger sættes
- PDB-filer indlæses
- Objekter og selektioner defineres
- Evt. alignments udføres
- Visualiseringer sættes op
- Orienteringen (synsvinklen) sættes
Gør nu følgende
4.1 Opret scriptet k-channel.pml
Opret et nyt script i din editor og gem det som k-channel.pml.
4.2 Initialisér scriptet
Start scriptet med at reinitialisere. Dette er rart, da man næsten altid kører scriptet mange gange under udviklingen og gerne vil have en “ren tavle” uden at genstarte PyMOL:
reinitialize4.3 Hent PDB-filen i scriptet
Skriv nu kommandoen til at hente PDB-filen:
fetch 1BL8, kchannel, async=0Her vælger vi at omdøbe objektet med det samme, så det hedder “kchanneli stedet for1BL8”. Det er en god idé, når man har mange strukturer og svært ved at huske dem fra hinanden. “async=0” sikrer at scriptet ikke kører videre før PDB-filen er helt indlæst.
4.4 Skjul standardvisning
Vi ved at PDB-filen kommer ind som cartoon som standard, så lad os fjerne det hele, så vi kan starte på en frisk:
hide all4.5 Opret selektioner for kæder
Skriv kommandoer til oprettelse af selektioner for hver kæde kaldet “chainA,chainB,chainCogchainD”.
4.6 Farv kæderne med color-kommandoen
Skriv kommandoer til at farve kæderne i forskellige farver vha. selektionerne, hertil benyttes kommandoen color, f.eks. vil
color tv_blue, my_selectionfarve selektionen my_selection blå. Du kan få inspiration til farver i Color-menuen, eller på denne side. Det er en god regel ikke at bruge de primære farver (red, blue, green, yellow, magenta, cyan), da de ikke kan repræsenteres korrekt på både skærm og papir. Det ser også mere professionelt ud at vælge nogle mere raffinerede farver, som f.eks. beryllium eller “gadolinium”. Du kan finde alle farver registret i PyMOL på følgende link; https://pymolwiki.org/index.php/Color_Values.
4.7 Vis strukturen som ribbon
Lad os nu vise hele strukturen som ribbon:
show ribbon, kchannelBemærk at kommandoerne show og hide benyttes til at gribe ned i menuerne til højre uden at klikke på dem.
4.8 Hent og indsæt set_view
Orientér nu strukturen som du gerne vil have den skal se ud (zoom, centrering, rotation). Brug venstre museknap til at rotere, højre museknap til at zoome og tryk på hjulet for at centrere. Skriv nu kommandoen (på kommandolinjen, ikke i scriptet):
get_viewPyMOL giver os nu en 3x6 matrix, der fuldstændig definerer synsvinklen, som det output der er vist nedenfor. Kopiér følgende,
set_view (\
0.649728715, -0.607225955, 0.457305670,\
-0.557611287, 0.028152177, 0.829624295,\
-0.516643345, -0.794027805, -0.320305407,\
0.000000000, 0.000000000, -203.462997437,\
72.186561584, 26.599575043, 19.889984131,\
164.716903687, 242.209091187, -20.000000000 )fra PyMOL og indsæt det nederst i dit script.
4.9 Fjern resterende selektion
Afhængig af hvordan, du har skrevet scriptet vil du muligvis opleve, at der vises en resterende selektion, når scriptet er kørt. For at undgå dette og fjerne den sidst valgte gruppe af atomer, indsæt kommandoen:
deselectIfm. TØ kan det være smart at gemme flere forskellige views eller repræsentationer af strukturen i såkaldte “scenes”, som let kan frembringes ved at klikke på knapperne nederst til venstre i PyMOL eller trykke F1, F2, F3...
For at gemme det nuværende view som en scene, bruges følgende kommando, som indsættes efter set_view-matricen ovenfor:
scene overview, storeDette gemmer den nuværende visning i en scene kaldet overview. Bemærk at man ikke kan bruge F-tasterne for at frembringe denne scene, da den er navngivet noget andet end F-. Man kan derfor i stedet enten trykke på den lille firkant med navnet overview i nedre venstre hjørne eller man kan skrive kommandoen; scene overview, recall.
4.10 Kør og test scriptet
Du har nu et komplet PyMOL-script, der først initialiserer, så henter PDB-filen, definerer selektioner og bruger dem og endelig sætter repræsentation og view. Test scriptet ved at kopiere hele teksten og indsætte den ind på PyMOL’s kommandolinje, enten øverst eller nederst, som angivet nedenfor. Tryk Enter/Return for at køre scriptet. 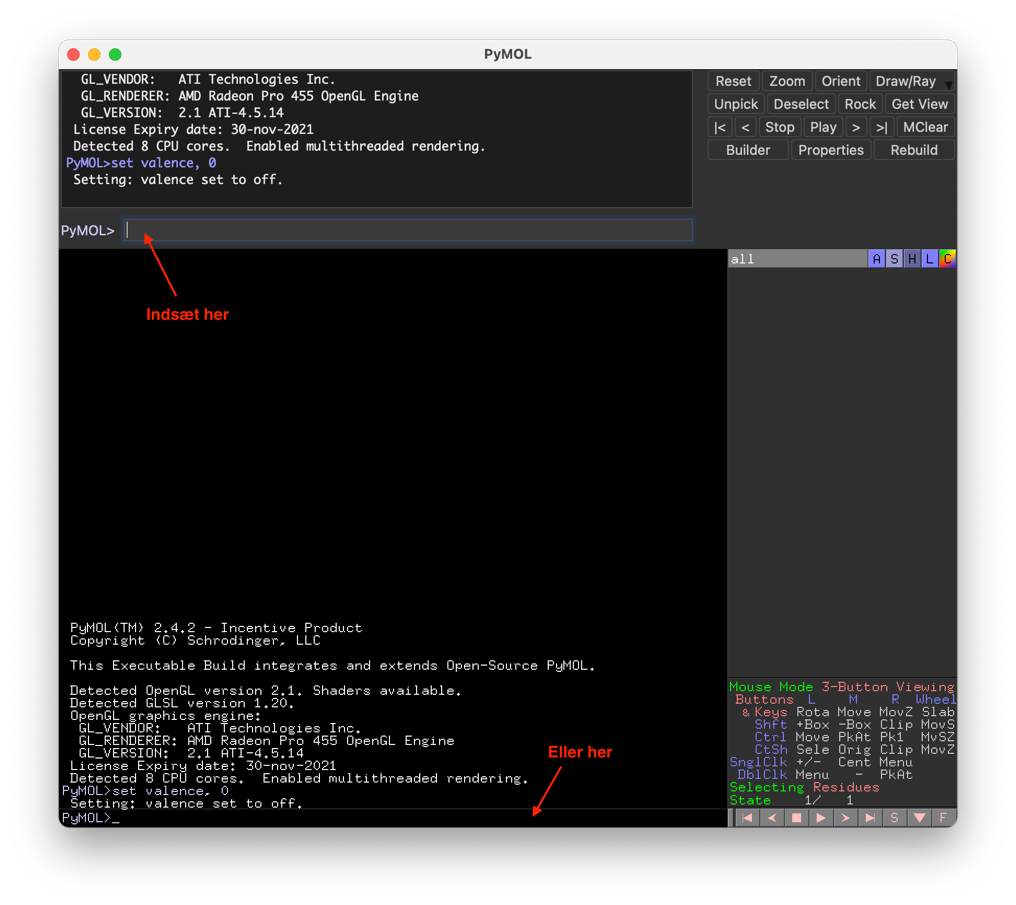
4.11 Orienter molekylet og test
Prøv at orientere molekylet på en anden måde og tjek at du kommer tilbage til start ved at klikke overview nederst.
4.12 Opret K+-ion selektion
Nu skal vi oprette et nyt view, der viser detaljer i K+-kanalens såkaldte selektivitetsfilter, der er med til at sikre, at kun K+-ioner kan passere. Denne del af scriptet skrives efter ovenstående og vil blive gemt i en ny scene. Opret først en ny selection med ionerne:
select k_ions, elem KDette udvælger alle atomer med grundstof K.
4.13 Vis ionerne som nb_spheres
Vis ionerne som non-bonded spheres (nb_spheres), der er mindre end alm. spheres, der viser VDW-radius:
show nb_spheres, k_ionsSæt også en passende farve på objektet efter frit valg.
4.14 Opret selektivitetsfilter selektion
Opret nu en selektion med kanalens selektivitetsfilter. Dette ligger i residues 75-78 i alle kæder, så vi kan nøjes med at skrive:
select filter, resi 75-78Vis filteret som sticks og find en god orientering, der viser filteret og ionerne oppefra. Indsæt orienteringen i scriptet.
4.15 Gem filter-scenen
Gem slutteligt den nye visning i scenen filter.
4.16 Hent overview-scenen igen
Scriptet skal vise overview først, så afslut af med at hente den ind igen:
scene overview, recall4.17 Kør og verificer scriptet
Kør det samlede script i PyMOL og check at der er to views, som man hurtigt kan skifte i mellem.
5 Opgave 5. Strukturel analyse i PyMOL
PyMOL scripting opgave: I vil i denne opgave blive introduceret for PyMOL-værktøjet Wizard measurement, som i senere vil kunne bruge til analyse af interaktioner.
Et vigtigt element i strukturel analyse er at kunne analysere interaktioner og detaljer i det aktive site eller mellem proteiner i et kompleks.
I denne opgave skal vi kigge på et eksempel, der er nævnt i kapitel 8 i Stryer, strukturen af cytochrome P450 bundet til substratet camphor (Fig. 8.5):
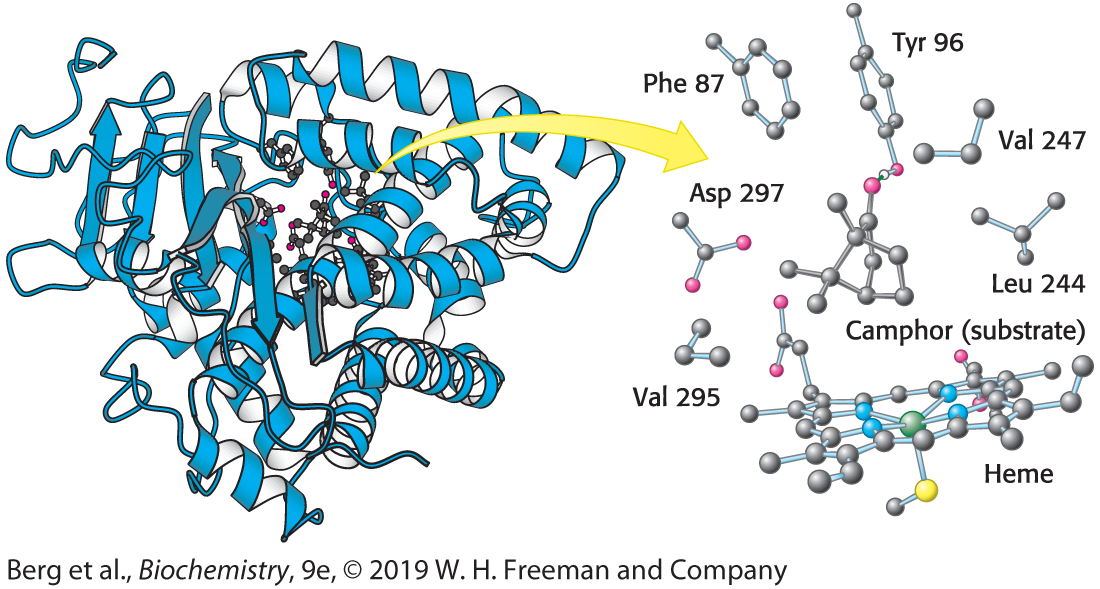
Vi vil gerne lave et PyMOL-script, der sætter os i stand til at foretage en grundig, strukturel analyse af hvordan camphor binder til proteiner, herunder hvilke typer af interaktioner (H-bindinger, Van der Waals- og hydrofobe interaktioner), der findes.
5.1 Hent og initialiser P450 scriptet
Start et ny PyMOL-script i VS Code med at reinitialisere og hent dernæst strukturen ovenfor via PDB-koden 2CPP, kald objektet cytochrome og skjul alle repræsentationer:
reinitialize
fetch 2CPP, cytochrome, async=0
hide all5.2 Vis strukturen som lyseblå cartoon
Vis hele strukturen i lyseblå cartoon (som ovenfor):
show cartoon, cytochrome
color skyblue, cytochrome5.3 Lav selektioner for hæm og camphor
Nu skal vi lave en selection med substratet, men vi ved ikke hvad det er kaldet i PDB-filen. Et godt tip til at finde ud af dette er at slå strukturen op på RCSB: https://www.rcsb.org/structure/2cpp. Under Small molecules kan man se at der både findes en hæme prostetisk gruppe (kaldet HEM) og liganden camphor (CAM). Disse tre-bogstavsforkortelse henviser til det, der står ud for “residue name” i PDB-filen, dvs. at vi skal bruge kommandoen “resn” til at vælge dem:
select heme, resn HEM
select camphor, resn CAM5.4 Lav selektion for active site
Vi vil også gerne have en selektion med alle de sidekæder, der interagerer med substratet, hvilket kan ses af figuren fra lærebogen:
select activesite, resi 87+96+297+247+244+2955.5 Vis selektioner som sticks
Nu viser vi alle tre selektioner som sticks:
show sticks, heme or camphor or activesiteBemærk brugen af den boolske operator OR her, som betyder at vi ønsker foreningsmængden af de tre selektioner, altså alle atomer, der ENTEN er i heme, i camphor, eller activesite.
5.6 Verificer aminosyrenummerering
Klik på et par af de udvalgte aminosyrerester for at forsikre dig at nummereringen i strukturen passer med lærebogen.
5.7 Tilpas activesite-selektionen
Kig nærmere på det aktive site, der er helt blåt. Bemærk at udover sidekæderne i det aktive site vises også hver aminosyres hovedkæde, hvilket vi ikke ønsker, da det forstyrrer når kun sidekæderne interagerer. For at fjerne disse må vi tilpasse udtrykket for activesite-selektionen. Ret derfor linjen ovenfor startende med select i dit script til følgende.
select activesite, resi 87+96+297+247+244+295 and (sidechain or name CA)Det tilføjede siger at vi ønsker alle atomer der opfylder at de befinder sig i de nævnte residues OG enten er sidekæde eller C-alpha (CA). PyMOL regner CA med til hovedkæden, så hvis vi ikke ønsker at sidekæderne skal flyve i fri luft er den en god idé at have det med.
Kør scriptet igen og tjek at alt fungerer som ønsket nu.
5.8 Farv hæm og camphor i scriptet
Nu vil vi gerne kigge på interaktioner, men de er lidt svære at se når alle atomer er blå. Vi kan dog ændre farverne for hæme og camphor samt benytte utility-funktionen CNC (Color Not Carbon), der farver alle atomer (pånær carbon) efter almindelige atomfarver. Den ligger i util-modulet og skal derfor kaldes på denne måde:
color palegreen, camphor
color firebrick, heme
util.cncBemærk at kommandoen ikke ændrede farven på carbon-atomerne i objektet og disse dermed bibeholder deres oprindelige farve.
5.9 Find passende view til active site
Indsæt nu en kommando så ingen atomer er valgt og find et passende view, der sætter dig i stand til at analysere interaktionerne i det aktive site. Husk at du kan bruge midterste museknap til at centrere, f.eks. på camphor, og hjulet til at skjule ikke-relevante dele af proteinet.
5.10 Gem oversigt og active site scener
Find også en god oversigtsorientering og gem de to som scener under overview og active site.
PyMOL er udstyret med en funktion til interaktivt at måle afstanden mellem to atomer i strukturen. Under fanebladet “Wizard” ligger værktøjet “Measurement”, der tillader brugeren at finde afstanden mellem to atomer i 3D-strukturen blot ved at klikke på først det ene og så det andet atom. Når der klikkes på et atom, anføres dette i logbogen øverst og et objekt ved navn measure## i objektoversigten til højre (hvor ## er nummeret på målingen). For at afslutte brugen af værktøjet klikkes på “Done” i nederste højre hjørne.*
5.11 Analysér interaktioner i active site
Brug nu active site-scenen til at analysere interaktionerne mellem cytochrome P450 og camphor/heme. Hvilke typer interaktioner findes der? Hint: Brug Wizard -> Measurement til at måle afstande.
6 Opgave 6. Mission Impossible i PyMOL
PyMOL scripting opgave: I denne opgave vil i få repeteret den scripting og de kommandoer i burde være bekendt med nu.
Denne sidste opgave er jeres svendeprøve i PyMOL-brug, de kommandoer I har lært samt scripting, inden I bliver sluppet løs på kurset. Hen ad vejen vil vi udbygge den viden, der er opbygget i denne indledende TØ, men lige nu er det vigtigt at få prøvet nogle af de ting af, du har lært. Det anbefales at opgaven laves i teams, hvor man diskuterer undervejs så alle forstår hvorfor scriptet skrues sammen som det gør.
Enzymet lysozyme (PDB-ID: 148L) findes i mange organismer, hvor det bruges til at angribe bakterier ved at kløve sukkerstoffer i deres cellevæg. Til opgaven skal I bruge strukturen af lysozyme kovalent bundet til et fragtment af cellevæggen i E. coli.
6.1 Slå lysozym op og identificer interaktioner
Slå strukturen op på www.rcsb.org og hent den ind i PyMOL.
Brug den indledende visning til at finde ud af hvilke kædenavne, der er brugt for lysozym og de to substratfragmenter. Tjek dette med hjemmesiden for at se om det stemmer overens.
Vælg H->water og S->lines for at vise detaljer for hele strukturen og skjule vand. Brug dette til at identificere (og nedskrive) en liste af aminosyrerester, der interagerer med substraterne.
6.2 Opret PyMOL-script til lysozym
Opret et PyMOL-script, der gør følgende:
Initialiserer og henter strukturen ind i programmet som
148L.Skjuler alt.
Opretter selektioner for lysozym og de to substrater.
Viser lysozym som cartoon i
violetpurple.Viser substraterne som sticks i to forskellige farver for kulstof og atomfarver for de andre atomer.
Opretter selektion for de rester, der interagerer og viser sidekæder for interaktionerne i violetpurple med atomfarver for andre end carbon.
Sørger for at ingen atomer er valgt.
Gemmer både en oversigts- og to detaljevisninger i tre scenes,
overviewsamtcell wallogsugar, der fokuserer hhv. på de to substratdele.
6.3 Analysér interaktioner med substraterne
Brug dit script til at analysere interaktionerne mellem lysozym og de to substrater. Hvilke typer interaktioner findes der? Hvilken aminosyrerest er kovalent forbundet til substrat?