Commands and scripting in PyMOL 3
0.1 Overview
To be an advanced user of PyMOL, who need to use commands and scripting. This tutorial provides a basic introduction to molecular visualization using commands and scripting in PyMOL 3. We will first introduce some general points about how best to work with the command line and then go through different usage examples that demonstrate basic functionality. At the end of the text we have provided a list of commands that can be used to look up useful commands.
0.2 Controling PyMOL on the command line and through scripting
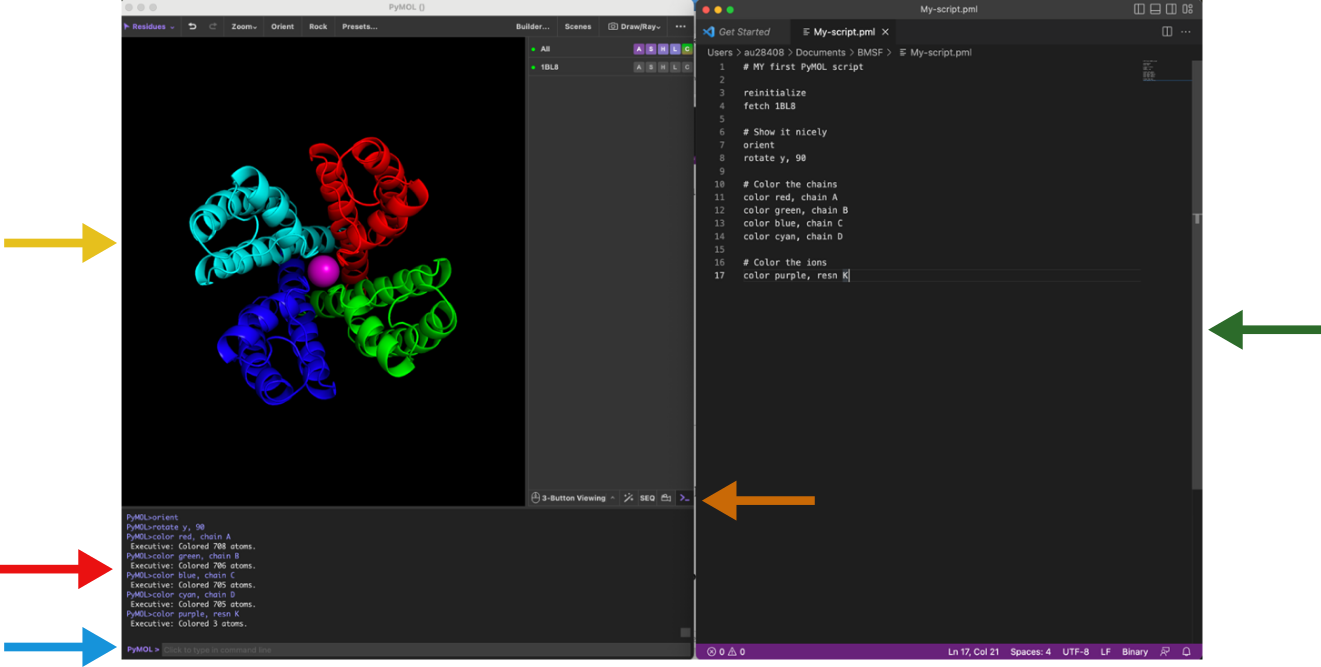
To work with commands and scripting you need to focus on the following windows and panels as shown in Figure 1. You need to think in input (the command/script) and output (the visualization). The command line is located at the bottom of the window and if you want a repository of commands, you can have them stored in a text editor on the side. The output visualization is seen in the main display panel. The log window contains log of which commands have been executed and details of selections. Note also the to hide/show the log window. A script is a list of commands in a text file that can be executed on the command line. A script helps you build up complex visualization and analysis pipelines that can be modified and reused.
0.3 Introducing the command line
All commands through the menu system can also be provided through the PyMOL command line console. You might find this complicated at first, but once you master it, you will find this very useful (if not essential).
To load a structure into PyMOL use the fetch command on the command line:
fetch 1rg7You can also load a PDB file from your own file system using the load command:
load 1rg7.pdbTo show a particular representation, use the show command followed the by representation name:
show cartoon (shows cartoon for all objects)
show lines (shows lines for all objects)To hide a representation use
hide linesYou can also choose to show only one particular representation with the as command:
as cartoon
as ribbonThe zoom and orient commands are the equivalent to the zoom and orient buttons, respectively:
zoom
orientWe will come back to more useful commands through the tutorial. Make sure you use
as linesbefore you continue to the next section.
0.4 Atom selections with the command line
Using the command line enables more versatile and specific atom selections. The “select” command enables atom selection through the command line. The command can be combined with various keywords specifying which atoms, residues, or chains to select. Here are some examples:
| Command | Description | Example |
|---|---|---|
select resname |
Selects the residue(s) by name. e.g. select residue with name MTX. | select resname MTX |
select resid |
Selects the residue(s) by identifier. e.g. select residue with ID 161. | select resid 161 |
select chain |
Selects atoms in the specified chain. e.g. select chain with ID A | select chain A |
select name |
Selects atoms with the specified name. e.g. select all CA atoms | select name CA |
The selection commands above will generate a selection with the default name (sele). You can specify another name by adding it to the command like this:
Create a new selection called mtx:
select mtx, resname MTXCreate a new selection called protein:
select protein, resid 1-160Similarly to using the in range (-) symbol, you can use the addition (+) syntax. The following command will select the residues in rage 15 to 20 as well as residue number 25:
select resid 15-20+25You can also combine selection statements using the AND and or operator. and will select the atoms present in both the first and the second statement (intersection) while or will select the atoms present in the first or the second statement (union). The following example will select 5 atoms corresponding to the CA (c-alph) atoms in the residues 15-20:
select resid 15-20 and name CARead more about atom selection in PyMOL here.
0.4.1 Exercise
Create a selection with name “bsite” consisting of residues with IDs 27, 31, and 94. Display the selection sticks representation on zoom on it.
0.5 Colour the atom selections
Now that we have a few selection entries (protein, mtx, bsite) we can easily color these to contrast the different elements in the structure.
Coloring can be done using the command line. Below we color the mtx selection red and the protein blue:
color red, mtx
color blue, proteinNote that it is often useful to color by atom elements, i.e. to keep oxygens red, nitrogens blue, sulphurs yellow etc, and only adjust the colors of carbon atoms. In the command line it can be carried out with combining the color command with selection statements:
color green, mtx and elem C color red, mtx and elem O
color blue, mtx and elem NNow we have coloured all carbon elements of the mtx selection green; oxygens red; and nitrogens blue.
1 Pymol Reference Card
1.1 Modes
Pymol supports two modes of input: point and click mode, and command line mode. The point and click allows you to quickly rotate the molecule(s) zoom in and out and change the clipping planes. The command line mode where commands are entered into the external GUI window supports all of the commands in the point and click mode but is more flexible and possibly useful for complex selection and command issuing. Commands entered on the command line are executed when you press the return key ⏎⏎.
| Description | Code |
|---|---|
| Command help | help <keyword> |
1.2 Loading Files
| Description | Code |
|---|---|
| File loading | load data/test/pept.pdb |
| Loading from terminal | pymol data/test/pept.pdb |
| Toggle between text and graphics | Esc |
| Toggle Y axis rocking | rock |
| Stereo view | stereo on/off |
| Stereo type | stereo crosseye / walleye / quadbuffer |
| Undo action | undo |
| Reset view | reset |
| Reinitialize PyMOL | reinitialize |
| Quit (force, even if unsaved) | quit |
1.3 Mouse Control
| Description | Code |
|---|---|
| Set the center of rotation | origin <selection> |
1.4 Atom Selection
| Description | Code |
|---|---|
| General selection syntax | object-name/segi-id/chain-id/resi-id/name-id |
| Molecular system selection | /pept |
| Molecule selection | /pept/lig |
| Chain selection | /pept/lig/a |
| Residue selection | /pept/lig/a/10 |
| Atom selection | /pept/lig/a/10/ca |
| Ranges | lig/a/10-12/ca |
| Ranges (multiple) | a/6+8/c+o |
| Missing selections | /pept//a |
| Naming a selection | select bb, name c+o+n+ca |
| Count atoms in a selection | count atoms bb |
| Remove atoms from a selection | remove resi 5 |
| General selections | all, none, hydro, hetatm, visible, present |
| Atoms not in a selection | select sidechains, ! bb |
| Atoms with a vdW gap < 3 Å | resi 6 around 3 |
| Atom centers with a gap < 1.0 Å | all near 1 of resi 6 |
| Atom centers within < 4.0 Å | all within 4 of resi 6 |
1.5 Basic Commands
Some commands used with atom selections. If you are unsure about the selection, click on the molecule part that you want in the viewing window and then look at the output line to see the selection.
| Description | Code |
|---|---|
| Fill viewer with selection | zoom /pept//a |
| Center a selection | center /pept//a |
| Colour a selection | colour pink, /pept//a |
| Force PyMOL to reapply colours | recolor |
| Set background colour | bg color white |
| vdW representation of selection | show spheres, 156/ca |
| Stick representation of selection | show sticks, a// |
| Line representation of selection | show lines, /pept |
| Ribbon representation of selection | show ribbon, /pept |
| Dot representation of selection | show dots, /pept |
| Mesh representation of selection | show mesh, /pept |
| Surface representation of selection | show surface, /pept |
| Nonbonded representation of selection | show nonbonded, /pept |
| Nonbonded sphere representation of selection | show nb spheres, /pept |
| Cartoon representation of selection | show cartoon, a// |
| Clear all | hide all |
| Rotate a selection | rotate <axis>, angle, <selection> |
| Translate a selection | translate [x,y,z], <selection> |
1.6 Cartoon Settings
Setting the value at the end to 0 forces the secondary structure to go through the CA position.
| Description | Code |
|---|---|
| Cylindrical helices | set cartoon cylindrical helices,1 |
| Fancy helices [tubular edge] | set cartoon fancy helices,1 |
| Flat sheets | set cartoon flat sheets,1 |
| Smooth loops | set cartoon smooth loops,1 |
| Find rings for cartoon | set cartoon ring finder,[1,2,3,4] |
| Ring mode | set cartoon ring mode,[1,2,3] |
| Nucleic acid mode | set nucleic acid mode,[0,1,2,3,4] |
| Cartoon sidechains | set cartoon side chain helper; rebuild |
| Primary colour | set cartoon color,blue |
| Secondary colour | set cartoon highlight color,grey |
| Limit colour to ss | set cartoon discrete colors,on |
| Cartoon transparency | set cartoon transparency,0.5 |
| Cartoon loop | cartoon loop, a// |
| Cartoon rectangular | cartoon rect, a// |
| Cartoon oval | cartoon oval, a// |
| Cartoon tubular | cartoon tube, a// |
| Cartoon arrow | cartoon arrow, a// |
| Cartoon dumbell | cartoon dumbell, a// |
| B-factor sausage | cartoon putty, a// |
1.7 Image Output
| Description | Code |
|---|---|
| Low resolution | ray |
| High resolution | ray 2000,2000 |
| Ultra-high resolution | ray 5000,5000 |
| Change the default size [pts] | viewport 640,480 |
| Image shadow control | set ray shadow,0 |
| Image fog control | set ray trace fog,0 |
| Image depth cue control | set depth cue,0 |
| Image antialiasing control | set antialias,1 |
| Export image as .png | png <image>.png |
1.8 Hydrogen Bonding
Draw bonds between atoms and label the residues that are involved.
| Description | Code |
|---|---|
| Draw a line between atoms | distance 542/oe1,538/ne |
| Set the line dash gap | set dash gap,0.09 |
| Set the line dash width | set dash width,3.0 |
| Set the line dash radius | set dash radius,0.0 |
| Set the line dash length | set dash length,0.15 |
| Set round dash ends | set dash round ends,on |
| Hide a label | hide labels, dist01 |
| Label a residue | label (542/oe1), "%s" %("E542") |
| Set label font | set label font id,4 |
| Set label colour | set label color,white |
1.9 Electrostatics
There are a number of ways to apply electrostatics in Pymol. The user can use GRASP to generate a map and then import it. Alternatively the user can use the APBS Pymol plugin. Pymol also has a built in function that is quick and dirty.
Generate electrostatic surface (built-in)
generate electrostatic surface action > generate > vacuum electrostatics > protein contact potential
1.10 Pymol Movies (mac)
| Description | Code |
|---|---|
| Move the camera | move x,10 |
| Turn the camera | turn x,90 |
| Play the movie | mplay |
| Stop the movie | mstop |
| Write out png files | mpng <prefix> [, first [, last]] |
| Show a particular frame | frame <number> |
| Move forward one frame | forward |
| Move back one frame | backwards |
| Go to the start of the movie | rewind |
| Go to the middle of the movie | middle |
| Go to the movie end | ending |
| Determine the current frame | get frame |
| Clear the movie cache | mclear |
| Execute a command in a frame | mdo 1, turn x,5; turn y,5; |
| Dump current movie commands | mdump |
| Reset the number of frames per second | meter_reset |
1.11 Miscellaneous
| Description | Code |
|---|---|
| Add hydrogens to a molecule selection | h add |
| Alias a set of commands | alias go,load 1hpv.pdb; zoom 200/; show sticks, 200/ around 8 |
| Structurally align | align prot1////CA, prot2, object=alignment |
| Fit one molecule to another | fit <selection>, <target> |
| Copy at selection | copy <target>, <source> |
| Create a new selection | create <target>, <selection> |
| Delete a selection | delete <selection> |
| Save file | save <filename>, <selection> |
| Protect or deprotect a selection | [de]protect <selection> |
| Mask or demask to allow/stop selection | [un]mask <selection> |
| Align coordinates with axis | orient <selection> |
| Get the current rotation matrix | get_view |
| Input a rotation matrix | set_view |
| Safely refresh the scene | refresh |
| Store a scene | view <name>, store, <description> |
| Restore a view | view <name>, [recall] |
| Set a new colour | set_color <name>, <rgb> |
1.12 Secondary Structures
Pymol has a secondary structure determination algorithm called dss, however it is better to use the DSSP algorithm and then define the limits manually.
| Description | Code |
|---|---|
| Run dss | dss <selection> |
| Define helical structure | alter 11-40/, ss='H' |
| Define loop regions | alter 40-50/, ss='L' |
| Define strand structure | alter 50-60/, ss='S' |
| Rebuild the cartoon after alteration | rebuild |
| Get dihedral angle | get dihedral 4/n,4/c,4/ca,4/cb |
1.13 Files
| Description | Code |
|---|---|
| Change the working directory | cd <path> |
| List contents of current directory | ls |
| Print current working directory | pwd |
1.14 Crystal Structures
To recreate crystal packing of molecules within 5 Å of pept in the pept.pdb (which must contain CRYST data), use the symexp command.
| Description | Code |
|---|---|
| Crystal packing within 5 Å | symexp sym,pept,(pept),5.0 |
1.15 NMR Structures
NMR models should be loaded into the same object, but should have different states.
| Description | Code |
|---|---|
| Load a model into an object | load <file>.pdb, <object> |
| Show all models in an object | set all states,1 |
| Show only one object model | set all states,0 |
| Show a particular model | frame <model_number> |
| Determine which model | get_model |
| Fit two structures to one another | fit <selection> |
| Fit and calculate the rms | rms <selection> |
| RMS without fitting | rms_cur <selection> |
| Fit ensemble structures | intra_fit <selection>,1 |
| Calculate rms | intra_rms <selection>,<state> |
| Ensemble rms without fitting | intra_rms_cur <selection>,<state> |
1.16 Changing Structures
| Description | Code |
|---|---|
| Add a bond | bond atom1, atom2 |
| Remove bonds | unbond atom1,atom2 |
| Join two molecules together | fuse [atom1, atom2] |
1.17 Old School Images
Load a .pdb, make cartoon view, set background to white then
| Description | Code |
|---|---|
| Change the ray mode to 2 | set ray trace mode,2 |
| Make the lines thinner | set antialias,2 |
| Raytrace the image | ray |